Fast and Accurate Whole Genome Indexing
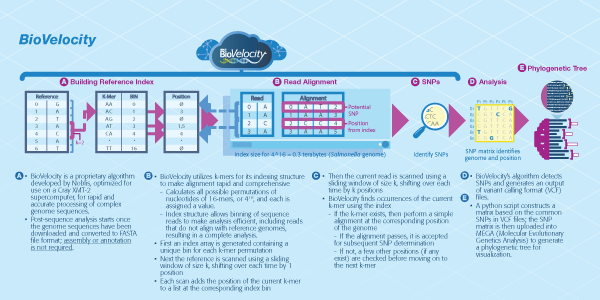
BioVelocity® is a bioinformatics tool based on an innovative algorithm and approach to genomic reference indices. Using a fast and accurate hashing algorithm, BioVelocity can quickly align reads to a set of references. BioVelocity takes advantage of a supercomputing system that is a scalable, massively multithreaded platform with a shared memory architecture optimized for large-scale data analysis and data mining—resulting in faster speeds, increased functionality, increased throughput and improved accuracy over current technologies. This supercomputing system enables us to use a brute force index, built out of all possible base pair sequences of various k-mer lengths. This index is used to map against thousands of references and allows for quick alignment of the k-mers amongst them simultaneously.
Noblis’ BioVelocity is a bioinformatics tool based on an innovative algorithm and approach to indexing genomic references. Using a fast and accurate hashing algorithm, BioVelocity can quickly align reads to a set of references and, through the use of high-performance computing platforms, produce faster results, increase functionality, increase throughput and improve accuracy in a bioinformatics workflow.
BioVelocity has a variety of functions:
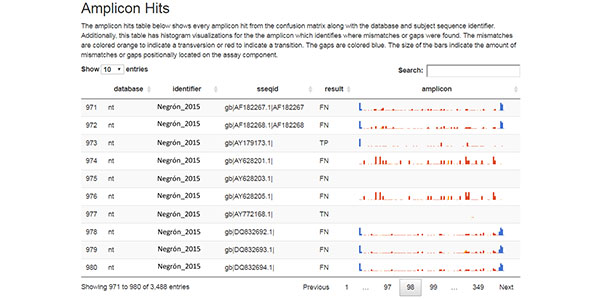
- Single nucleotide polymorphism (SNP) detection – SNPs are a type of genetic variation and each one represents a difference in a single DNA building block, called a nucleotide
- Metagenomics analysis – the application of bioinformatics tools to study the genetic material from environmental samples without first culturing the present microorganisms
- Conserved and signature sequence detection and compression - rapidly reduce the dataset of two bins of target sequences: (1) those that are conserved between the target organism and the reference genomes and (2) those that are unique to the target organism compared to the reference genomes
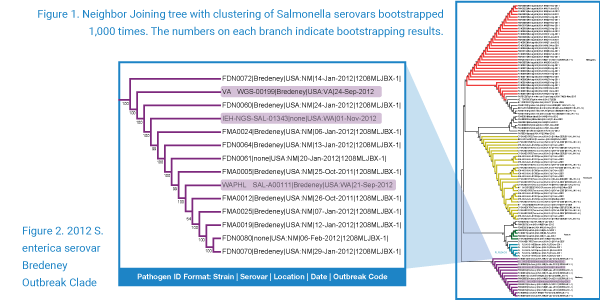
BioVelocity provides insight into the molecular mechanisms of pathogen evolution, virulence, host preference, lineage calculations and the emergence of highly pathogenic strains via advanced SNP detection algorithms. Advances in sequencing technology have increased the computational demands required for processing, identifying and analyzing large, complex datasets. When it comes to dangerous pathogens, speed is critical, and BioVelocity delivers.
Want to know more about how BioVelocity works? Click here.